Study Title: Pramipexole Trial for Bipolar Depression - PAX-BD
A randomised, double-blind, placebo controlled trial of pramipexole in addition to mood stabilisers for patients with treatment resistant bipolar depression.
Chief Investigator: Professor Hamish McAllister-Williams
This document provides a summary of the PAX-BD study and what it involves for sites taking part. If
your site would like to take part and you have not already completed a feasibility questionnaire,
please do so here https://www.smartsurvey.co.uk/s/PAXBDv2/ this will be sent directly to the Trial
Managers.
To contact the Trial Managers directly, please email PAX.BD@newcastle.ac.uk
Benefits for sites taking part in PAX-BD:
- PAX-BD is an NIHR funded trial and eligible for portfolio adoption and service support
- PAX-BD is supported by two dedicated Research Assistants who will be in regular contact with the participants and site teams, monitoring the online mood scorings and feeding information back to local clinicians.
- Local clinical staff will have the opportunity to gain research experience as well contributing to increasing the evidence base for the management of bipolar disorder.
- Local staff will gain increased expertise in managing bipolar disorder via guidance provided by senior study co‐applicants: Profs Allan Young (London), John Geddes (Oxford), Richard Morriss (Nottingham), Hamish McAllister‐Williams (Newcastle), Danny Smith (Glasgow)
- All sites will be given a clinician manual with detailed clinical guidance in the use of pramipexole and for managing patients taking part in PAX-BD.
- Recruiting to PAX-BD will increase the local Trust’s research activity.
- Sites will receive money towards their set up as well as a per patient recruitment amount.
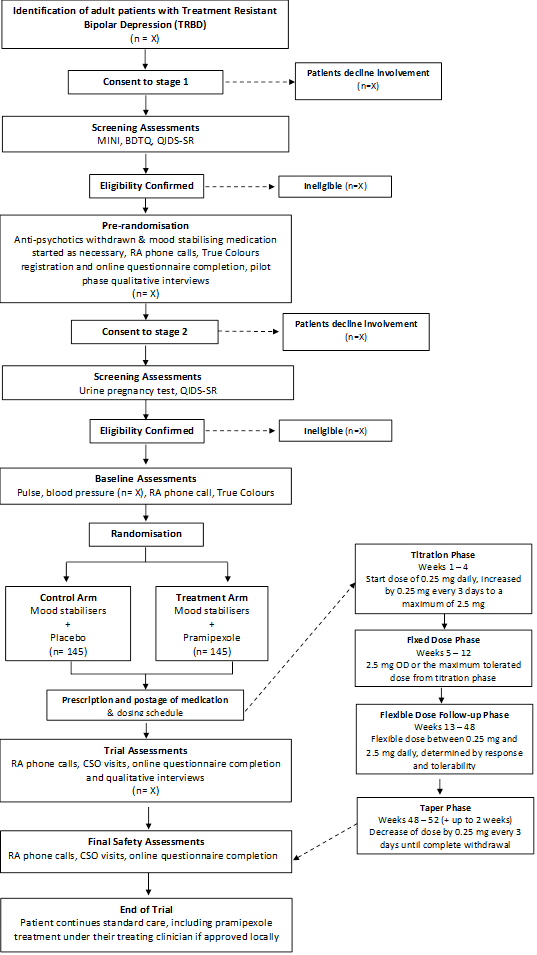
Process flow diagram
TRIAL SUMMARY
This document provides a provisional SUMMARY of the trial. It is not a substitute for the trial protocol, which will include full and final details of trial conduct at sites.
Study population
Adults with Treatment-resistant Bipolar Depression (TRBD). This is defined as failure of two NICE-recommended medications.
Pre-randomisation Phase
‘Stage 1’
A study visit will be arranged to receive written informed consent and determine if the patient is eligible to for stage 1.
After patient consent, the PI or local delegate will administer the following assessments in order to assess eligibility for stage 1, as a minimum:
- Medical history and assessment of diagnosis/diagnoses according to DSM-5 criteria, assisted by the use of the MINI
- Bipolar Demographics and Treatment Questionnaire
- QIDS-SR (paper)
Patients will have their antipsychotics withdrawn and mood stabiliser initiated if necessary. Stage 1 will determine if patients are able to adhere to weekly online mood ratings and engaging in telephone calls with the Research Assistants (RAs).
All participants must have been on mood stabilising medication for a minimum of four weeks before being assessed for stage 2.
Sites will be given a Clinician Manual, with detailed information and trial SOPS.
Randomisation Phase
‘Stage 2’
A screening and randomisation visit will be arranged to receive written informed consent and determine if the patient is eligible to be randomised to stage 2.
After patient consent, the PI or local delegate will administer the following assessments in order to assess eligibility for randomisation, as a minimum:
- QIDS-SR (paper)
- A urine sample pregnancy test for all female participants of child-bearing potential.
Trial clinicians will be advised to arrange for other pre-treatment tests that they consider to be necessary for the safety of their patients.
Once confirmed as eligible, the PI or delegated clinician will communicate this with the patient and these eligible participants will be randomised anonymously (double blind) to receive either pramipexole (active treatment) or placebo.
Trial summary (cont)
Trial Medication
Prescriptions for trial medication must be written by a member of staff delegated to do so on the site delegation log and sent to sponsor pharmacy. All trial medication will be sent directly from sponsor pharmacy to the participant home address by tracked and signed delivery. If a participant stops taking medication at any point during the trial, a taper and stop prescription may be required to ensure the participant has sufficient quantities of both strengths of medication to taper dose gradually.
Phase 1: Titration phase (Weeks 1-4)
Participants will start at a dose of 0.25mg daily which will increase by 0.25mg every 3 days to a maximum dosage of 2.5mg if tolerated by end of Week 4.
Phase 2: Fixed dose phase (Weeks 5-12)
The dose attained at the end of Week 4 is continued.
Phase 3: Flexible dose follow-up phase (Weeks 13-48)
Beyond 12 weeks, pramipexole will be flexibly dosed between 0.25 and 2.5mg/day, determined by response and tolerability and guided by the algorithm in the protocol.
Phase 4: Taper Phase (Weeks 48-52, + up to 2 weeks)
At the end of the trial or if a patient needs to stop medication for any reason, medication will be tapered. Trial medication must never be stopped suddenly. Dose reductions during tapering will be made at a rate of 0.25mg every 3 days. Clinicians will receive further guidance for tapering as part of the Clinician Manual.
Total recruitment duration
22-month recruitment period, in total.
Trial summary (cont)
Follow-up
Participants will be followed up to 12 months (52 weeks +up to 2 weeks)
Sample size
290 participants to be randomised to the intervention phase
Site recruitment target
To be agreed with the sponsor, CI and the Newcastle Clinical Trials Unit.
Participating sites
40 Trusts.
Additional tests
Pregnancy test prior to randomisation.
Data collection and recording
There will be a small amount of data entry required at sites, for example some screening information. The majority of data will be entered by participants or collected by the RAs.
Outcome data, in the form of answers to all trial questionnaires (Health Economic and otherwise) will be entered directly by the trial participants into the TrueColours online platform. Participants will receive login details at the start of the trial. They will also receive reminders to complete the questionnaires throughout the trial as well as telephone support by the trial RAs.
Research Assistants (RAs)
Two trial Research Assistants will be in regular contact with participants by telephone at specific times during the trial. This will include addressing issues and missing data on TrueColours, prompting participants to complete their data, checking and recording adverse events and side effects, and checking and recording IMP compliance.
They will check that participants are taking their medication, and forward any queries about dose increase/decrease to the PI as appropriate.
Staff resource required at site
- 1 Local PI per site
- Medical Delegate(s)
- CSO/equivalent site staff member – 7 visits during stage 2 to take pulse and blood pressure, and to collect remaining/unused trial medication. If you need additional support for the CSO visits and data entry that cannot be found within your Trust, you can approach your local Clinical Research Network (CRN). The 15 local CRNs in England have been informed of the study and that requests for support may be sent to them, and discussions are taking place with the CRNs in the devolved nations.
Note: consent and confirmation of eligibility must be undertaken by the PI or delegated (GMC registered) clinician on the delegation log.
Finances
£300 to cover study set up
£69.88 per randomised patient
These amounts are currently being confirmed however the final numbers will be included in your site agreement as part of your R&D review.
ETCs for pramipexole are to be claimed through your local Clinical Research Network and reimbursed to NTW (who have paid for the trial medication for central distribution to participants). Details for reimbursement will be included in your site agreement. Eligibility Criteria
Always refer to the current approved trial protocol. The below may be subject to change. All
participating sites will receive training in the protocol and applicable regulatory processes from
members of the Trial Team.
Patients must fulfil all of the following criteria to enter the pre-randomisation phase:
STAGE 1: PRE-RANDOMISATION
| Inclusion Criteria |
| 1. Currently under the care of secondary care mental health services at screening with a plan for the patient to remain in secondary care throughout the period of the trial. |
| 2. A decision made by the patient’s clinical team that a change in medication is indicated. |
| 3. A current diagnosis of Bipolar Disorder (type I or II), defined as in DSM-5, which is supported by the use of the Mini-International Neuropsychiatric Interview (MINI) (55). |
| 4. Currently depressed, i.e. meeting DSM-5 criteria for a Major Depressive Episode assessed via MINI and with a current QIDS-SR >10. |
| 5. Current episode of depression failed to have responded to adequate trials, or lack of tolerability or patient refusal, of two different NICE recommended medications (quetiapine, olanzapine + fluoxetine, lamotrigine) or lurasidone. Adequacy of treatment trial defined using a custom designed ‘Bipolar Demographics and Treatment Questionnaire’ (BDTQ). |
| 6. Aged 18 or over at the point of consent. |
| 7. Willing and able to provide written informed consent prior to any trial procedures taking place. |
| 8. In the opinion of the investigator, is able to follow the trial prescription instructions and is able to manage 8 weeks supply of trial medication without risk of overdose. |
| 9. For females of childbearing potential: Willing to use highly effective contraception or to practise true abstinence to avoid pregnancy during the pre-randomisation and post-randomisation phase of the trial. |
STAGE 1: PRE-RANDOMISATION
| Exclusion criteria |
| 1. DSM-5 defined severe substance use disorder. |
| 2. Current psychotic symptoms as assessed using the MINI. |
| 3. History of retinal disease. |
| 4. Current cardiovascular symptoms or significant concerns around cardiovascular disease. |
| 5. History of renal disease. |
| 6. Any known sensitivity to trial drug including its excipients. |
| 7. Current pregnancy or planned pregnancy during the trial period, or breastfeeding |
| 8. Starting specific psychotherapy from four weeks before randomisation through to Week 12 post-randomisation. |
| 9. Currently taking part in another clinical trial that would interfere with the outcomes of PAX-BD (site team to check with the CI and Trial Management Group if in doubt). |
| 10. Confirmed diagnosis with potential confounding factors such as Parkinson’s disease, restless leg syndrome. |
| 11. Clinical concern of previous impulse control behaviours including harmful alcohol or drug use, binge eating, gambling or sexual behaviours, or regarding significant suicidal risks. |
STAGE 2: AT RANDOMISATION
| Inclusion criteria |
| 1. Currently depressed, i.e. meeting DSM-5 (56) criteria for a Major Depressive Episode and with a current QIDS-SR >10. |
| 2. A minimum of two telephone phone calls with a trial RA and two on-line weekly symptom ratings have been completed during the pre-randomisation phase |
| 3. On mood stabilising medication (lithium, valproate, carbamazepine, lamotrigine) |
| 4. All regular psychotropic medication, including mood stabilisers, at a stable dose for a minimum of four weeks |
| 5. The patient, if female and of childbearing potential, must have a negative pregnancy test [urine beta-human chorionic gonadotropin (β-hCG)]. |
6. Sexually active female patients of childbearing potential are required to practice true abstinence in line with their preferred and usual lifestyle or use a highly effective contraceptive method for the treatment duration. Highly effective methods of contraception include:
- combined hormonal contraception associated with inhibition of ovulation (oral, intravaginal, transdermal)
- progestogen only hormonal contraception associated with inhibition of ovulation (oral, injectable, implantable)
- intrauterine device (IUD)
- intrauterine hormone-releasing system (IUS)
- vasectomised partner
- bilateral tubal occlusion
|
| 7. Willing and able to confirm written informed consent at the point of randomisation, after the pre-randomisation period. |
STAGE 2: AT RANDOMISATION
| Exclusion criteria |
| 1. Psychotic symptoms over the preceding 4 weeks. |
| 2. Any known sensitivity to trial drug including its excipients |
| 3. Any deterioration in physical or mental health since pre-randomisation that means there is a clinical concern to proceed with the study. |
| 4. On an antipsychotic at the point of randomisation. |
| 5. Current or planned pregnancy during the trial period, or breast feeding. |
| 6. Starting specific psychotherapy from four weeks before randomisation through to Week 12 post-randomisation. |
| 7. Currently taking part in another clinical trial that would interfere with the outcomes of PAX-BD (site team to check with the CI and Trial Management Group if in doubt). |
| 8. Confirmed diagnosis with potential confounding factors such as Parkinson’s disease, restless leg syndrome. |
| 9. Clinical concern of previous impulse control behaviours including harmful alcohol or drug use, binge eating, gambling or sexual behaviours or regarding significant suicidal risks. |
| 10. Any study team’s concern regarding the patient’s ability to remain engaged in the study collecting self-ratings of their symptoms. |
This question requires an answer
Full organisation postal address and postcode:
Please ensure you complete your contact details